简 述
合理的实验模型永远是过程开发的主题。复杂反应的基本特征是过程产生多种产物,而目标产物通常先于其他产物出现,在后续过程中转换成价值很低甚至没有价值而成为负担的副产物。因此,需要研究每个特定过程的机制,设计出恰当的系统和实验模型。
一个产品的实验模型可以有多种猜测,本文以有机硅直接合成为例,给出一些与现有实验模型不同的复杂气固催化过程的实验模型,供有关领域专家参考。
1.前言
氯化氢、氯甲烷或某些简单醇与硅在催化剂存在条件下合成氯硅烷、甲基氯硅烷和烷氧基硅烷的反应,
在幸松民,王一璐《有机硅合成工艺及其应用》一书中有详细的综述。
在铜催化剂及高沸点有机介质存在的条件下,硅与一元醇 ROH (R为烷基)在高温下反应合成烷氧基硅烷,
和从氯硅烷出发醇解的工艺路线相比有工艺简单、几乎没有有害副产物、
没有氯气腐蚀设备的问题和成本低的特点而受到高度重视。
目前的技术具有如下的模型(取自《有机硅合成工艺及其应用》):
按照这个猜想的指导来设计系统,只能是
全混反应模式。典型的实验结果是粗产物中甲醇三分之一,三甲氧基硅烷三分之一,四甲氧基硅烷等无用组分也有三分之一或更多。
硅与碳同族,为四价元素。类比推定,在硅与甲醇反应过程中,烷氧基硅烷至少有五种产物,
通式为 HnSi(OR)4-n(n=0,1,2,3,4) ,目的产物通常为 n=1,2。
因此,醇与硅的反应过程不属简单反应,而是复杂反应,存在选择性问题。
我们首先作宏观分析,探讨实验模型。
2 醇与硅反应的热力学
首先要解决的问题是反应能否发生。热力学有基本的算法,
由标准状态下反应的吉普斯自由能变化值 ΔrGθm,可以大体估计反应发生的可能性。
ΔrGθm 由下式估计(详见《物理化学》及 CRC 化学手册):
ΔrGθm(Tθ)=ΔrHθm
-TΔrSθm ------(2.1)
若化学反应是在标准压力 Pθ 和标准温度Tθ 下进行的,则
ΔrSθm(Tθ)=∑νBSθm(B,Tθ)
------(2.2)
压力为 Pθ 时,任意温度下化学反应的熵变为:
所需要的基础热力学数据不能都找到,不能完成这些计算。因此只能用实验的方法来进行观察和分析。
上述系统运行得到了三甲氧基硅烷、四甲氧基硅烷等多种产物。
粗产物中如果含有甲醇,即使在很低的温度下,贮存期间三甲氧基硅烷也会逐渐减少,
而不期望的产物四甲氧基硅烷在逐渐增加。可以推断,
三甲氧基硅烷很活泼,不需要催化剂存在就能与甲醇发生反应变成四甲氧基硅烷。即,下一反应存在且不可逆,
HB(OR)3 + ROH ⟶ B(OR)4 + H2 ------(2.4)
简而言之,烷氧基硅烷上的氢可以被甲氧基取代,反过来则不行。
实践还发现,在蒸馏过程中,甲醇与三甲氧基硅烷共沸,提纯难度很大。
即使很少量的甲醇进入产品中,产品的贮存和应用就会产生很大的问题。
除发生上述副反应外,有些副反应产水,水又带来一系列副反应。
在反应过程中,水解反应使硅烷变成不溶不熔的产物堵塞系统。
在贮存过程中,使产品迅速变质。目前市场上产品质量保证期标定为 6 个月。
物化手册没有查到二甲氧基硅和一甲氧基硅的资料,可以猜想,
二甲氧基硅烷和一甲氧基硅烷,比三甲更加活泼,不能或很难单独存在。
这些产物是怎么产生的?热力学,动力学和统计学各有不同的说法。我们暂不讨论这些纯粹理论性的问题。
在宏观上,我们可以假设一甲、二甲和三甲都可以在反应的第一时间发生。
产生之后,只要其周围存在甲醇,就会以它们各自的速率继续反应,氢被甲氧基取代。
如果不加任何干预,不采取恰当措施,最终连三甲氧基硅也会变成四甲等不含氢键的产物。
这些转换的条件是反应产物与原料醇频繁的返混,在返混过程中,含氢键较多的物质其氢键陆续被甲氧基取代。
因此,可以进一步推断,全混流反应器不是这类反应的最佳选择。
3. 甲醇的解离与碰撞统计
甲醇有两种键断裂方式。用 R 代记甲基 (CH3)。在一般条件下,25 ℃时,R-OH 断裂能为 387kj/mol,
RO-H 断裂能为436kj/mol,前者较容易断裂。在特定条件下,可以使 RO-H 断裂方式相对于 R-OH 断裂方式发生的机率更大。
这样考虑,其离解后的基团可能有四种:R/OR/OH/H。产物具有结构形式
HaSiRbORcOHd, (a+b+c+d=4, 0≤a,b,c,d ≤4)------(3.1)
進一步假定(R-OH)断键方式的概率为 P
1,(RO-H)断键方式的概率为 P
2, (P
1+P
2=1) 。
则 n 个醇分子离解后产生出 nP
1个 R
- 和 nP
1 个 OH
+, nP
2 个 OR
- 和 nP
2 个 H
+。
把 nP
1简记为 n
1,nP
2 简记为 n
2,则 H
aSiR
bOR
cOH
d 出现的概率为:
P= Can1Cbn2Ccn1Cdn2 / C42n------(3.2)
全部可能的产物共有 35 种。在醇硅直接合成过程中的产物的色谱分析图上可以查到 30 余个特征峰就是证据。
产物出现概率的较好的估计需要这些离子的离子半径,准确数据尚未找到。
解离方式有四种可能:
- 极性解离;
- 催化解离;
- 吸附解离。
- 不发生解离。
笔者现有的认识,本反应中的甲醇需要解离,三种解离方式都可能存在,而溶剂的极性解离作用最大。
因此,极性溶剂是必要的。溶剂除了起悬浮作用之外,最重要的作用是实现和加速甲醇的解离。
如果甲醇不能快速解离,反应不能快速按指定方向进行。
假设某种催化系统能抑制 R-OH 断裂并提高 RO-H 键的断裂概率,这种假设没有坏处。
离开催化剂,本反应几乎不发生。
4. 醇-硅过程的均相反应模型
要设计反应系统,必须设想其反应原理模型,哪怕是简化的,宏观的。
为推导与编程方便,将硅简记作 B1(不致误会时它就是 B),
H2B(OR)2 为 B2, HB(OR)3 为 B3,
B(OR)4 为 B4, RB(OR)3 为 B5 。猜想主要存在如下五个反应:
建立微分方程并求解,可以得到一组方程,(微分方程的建立与求解过程及解组放在后面的附录中。)
编写计算代码,假设一组状态参数(反应速率 ki)模拟运行。
运行图如图 4.1 所示。
图 4.1 醇-硅过程模拟运行图
全混流反应模型本质上就是这个模式。一个是固体,一个是气体,醇-硅过程当然不是均相的。但是,我们可以借用这个模型了解过程的某些特性。
在某一试验阶段使用这一模型了解过程的基本特性是必要的。后面我们再讨论如何修正这一实验模型。
图 4.1 中的醇指数是 t 时刻 各组分(Bi)所含甲基之和摩尔数计算值,
2B2+3B3+4B4+4B5。
它与醇的实际供给量(图中左下角至右上角的斜线)不重合,醇的缺失是假的,因为这些速率的模拟是随意假设的而不是测定的。
另一方面,这一现象反映过程的本质。有一个时刻,在这个时刻之前,醇处于饥饿状态,
反应处于激烈的竞争中。之后,竞争相对不激烈,以致逐渐消失。
由图 4.1 很清楚地看出,反应(4.1)-(4.5)是复杂反应,可以分为三组:
第一组:反应主要为(4.1)的引发生成 B2;
第二组:平行反应(4.1)、(4.3)竞争地发生,消耗醇和 B2 分别生成 B3 和 B4;
第三组: B3 出现之后,平行反应(4.4)、(4.5})发生,又竞争消耗醇和 B3 ,分别生成 B4 和 B5,
组间具有串反应性质。一旦 B2,B3 出现之后,这三组之间又互相竞争。竞争消耗醇和目标产物。组间已失去时序关系,发生恶性竞争。
存在一个时刻 t, B2 达到最大值, B3 随后达到最大值。B3 一旦出现, B4,B5 即获得出现的可能,
逐渐出现并迅速增加且不减。一旦 B5出现,就意味着有水出现了,系统将面临各种麻烦。
反应的控制在于醇。只要有醇,即使没有硅了,反应照样能够继续,直到全部组分变成 B4或 B5。
如果没有醇,即使有再多的硅,反应会立即停止,各种产物的浓度不会继续发生变化。
假定在某一时刻,停止加入醇。在系统逐渐消耗完剩余的甲醇之后,反应会停止。
过程开发的要求是使主产物浓度最高而副产物浓度较低。
其措施只能是选择一个恰当的时刻,停止加入醇使反应停止。将目标产物提取出来后再进入下一反应周期。
5. 系统的体积特性
若真如 (4.1)-(4.5) 所示,在稳态条件下,反应 (4.1) 使体积减半,而其余反应不改变系统的体积。
正常情况下,除了硅,气体由硅烷,氢气组成。即,被转换的醇,其体积一定会减少一半,变成了硅烷和氢气。
假如 M 摩尔醇都被转化为硅烷,则流通的体积减少一半。如果流动体积不变,反应没有发生。
如果尾气含氢,则标志着(4.2)-(4.4)发生了。氢气很少,转换方向不利。氢愈多,反应愈强烈。
系统堵塞故障由反应 (4.5) 所致。
刘清源教授指教笔者,氯化亚铜是甲醇分解催化剂,在高温下,反应的主要倾向是。
一个体积甲醇分解得到三体积的气体。在稳态条件下,气体流速增加两倍,检测尾气成分可以做出判断。
6.三相反应模式
在甲醇与硅合成烷氧基硅烷的直接合成技术中,大多数专利采用三相反应模式。在反应器中加入高沸点溶剂,
同时存在气-液-固三种形态。
在这个过程中,溶剂到底起什么作用,是个值得探讨的问题。有人称之为悬浮剂,因为溶剂的存在,
固体物借溶剂得到浮力,因而有利于搅拌,有利于固体物料的分散和传质传热。
全混间歇反应方法借助机械作用使固体物运动,在运动中与气相反应物充分接触。实现气固反应。
溶剂的存在,减轻固体物料对搅拌器的磨损。
反应 (5.1) 表明,在甲醇与硅合成烷氧基硅烷的直接合成过程中,若以氯化亚铜作催化剂,必须有极性溶剂存在,起着极性解离作用。
否则反应不会倾向烷氧基硅烷方向,在高温下的倾向是分解,(4.1)-(4.5) 不会发生。
因此,溶剂的最重要的作用是所谓溶剂化效应,使醇解离而不是分解。
由此,使醇按指定模式解离是提高选择性的关键,解离的速度制约反应速度。溶剂的设计和选择很重要。
三相反应技术有一些致命的弱点。溶剂占据了一定的反应器空间。
高纯度高沸点溶剂十分昂贵,带来了新的杂质源和新的成本要素。
如果溶剂不起解离作用,其存在必起隔离作用,使气相反应物和固体颗粒隔离开来。
固体颗粒周围增加了新物质,氛围更加复杂,增加了气体扩散的阻力。
原料气在溶剂中形成气泡,必须有高速搅拌来使系统均相化。
搅拌速度的提高又加大了固体颗粒与搅拌器之间的摩擦力,也使液相乳化,甚至需要加入抗乳化剂,系统越来越复杂。
溶剂的应用也必定制约生产规模。这些问题促使人们寻找摆脱溶剂的方法。
是否存在某个条件,不用溶剂,产出三甲氧基硅烷,有待研究。
摆脱溶剂的关键是保证醇的解离模式为 RO-H 键断裂,没有了溶剂的极性,就必须寻求特殊的催化剂或某种其他技术。
7. 化学吸附
在反应进行条件下,甲醇及 Bi(i=2,3,4,5) 是气相物,而 Si 是固体粉末。
因此,反应(4.2)-(4.5})可以看作是气-液或气-气反应,反应(4.1)则是气固非均相的。
每个硅原子地位不可能平等,只有与甲醇发生接触的被激活硅的原子有机会参与反应。上述第四节的均相反应模型需要修正。
根据物理化学和化学反应工程的一般原理,当只考虑反应(4.1)时,过程包括以下 4 个步骤:
- 原料气向固体表面扩散;
- 原料气被固相物料吸附;
- 在活性中心处进行化学反应;
- 产物气从固相物料表面解吸进入气体物流中。
这个过程叫做化学吸附。它一定会经历扩散、吸附、化学反应和产物解吸四个过程,对应耗去四个在宏观尺度上很小的时间片段,
t
扩散, t
吸附, t
反应, t
解吸 四段时间,称为一个吸附周期。
这些量是些统计量。硅与醇的化学吸附可以想象为解离化学吸附。醇按需要解离,否则难以离解得到所需的产物。
但是,甲醇究竟是由于硅的表面吸附作用解离,还是溶剂的极化作用而解离或者二者兼有?不同的解离机理其扩散方式不一样。
吸附解离的扩散是甲醇的扩散,解离发生在硅粉表面和空隙中。溶剂极性化作用的解离发生在溶剂中,扩散是未解离的醇与解离出的离子的扩散。
这二者对反应的控制方式不同,加速反应的措施不同,实验模型不同。
8. 缩芯模型
黄恩才在《化学反应工程》一书中引用 Yagi 和 Kunii 的理论,把固体颗粒直径逐渐缩小的反应过程称为缩芯过程。
每一个固体颗粒被一层气体滞流膜包裹着。扩散是指原料气透过这层气体滞流膜向颗粒表面扩散。
用这一理论来解释甲醇与硅的气固过程,反应发生在硅颗粒的表面,硅粉颗粒粒径在过程中逐渐变小并最后消失。
如果硅粉不含非硅固体杂质,不会留下灰分。
甲醇通过一个颗粒半径为 Rs,膜半径为 Rg 的气体滞流膜向内扩散,速率与颗粒表面积 S 和甲醇浓度差成正比
-dnM/dt=kgS(CMg-CMs) ------( 8.1)
式中 k
g为传质速率, C
Mg 为滞流膜外界面处的醇浓度,C
M为固体颗粒表面的醇浓度。
在颗粒表面上发生化学反应的速率,即甲醇的分子数消失速率与硅颗粒的表面积和颗粒表面处的甲醇浓度的平方成正比
-dnM/dt=kSC2Ms ------( 8.2)
式中 k 为化学反应速率。令 b=k
g/k,联立 (8.1),(8.2) 二式,可以得到
CMs={ -b±√[b2+4bCMg]}/2 ------( 8.3)
C
Ms 取正值和合理值。将此式代入 (8.2) 式,即得甲醇分子数的消失速率。
(8.2)式当然也可以改换视角,硅颗粒的分子数消失速率为醇消失速率的一半
-dnSi/dt=-dnM/2dt=kSC2Ms/2 ------(8.4)
这里含有三个工程量,S,k
g,k 都是时间的未知函数。
单位表面积上的硅原子数可以看作是一个常数,因而颗粒表面的硅原子总数与总面积成正比。
nSi∝ S ,把比例常数并入反应速率常数中,(8.4) 式右边表达式成为 0.5knSi
C2Ms。
该式指的是硅原子的消失速率,也就是 B2 分子产生的速率。除了表达方式之外,与(4.1)式没有本质的区别。
令 Bi 表示各组分的分子数,则两个模型的表述完全一致。不难得到分子数量与浓度之间的关系。
由 (8.1) 式,很自然地得到推论,固体反应物的颗粒越细则总表面积越大,反应越快。
表面积与粒半径和物质密度 ρ 成反比。
S=3/(rρ)
因此,硅粉颗粒应当细。硅的消耗与颗粒表面上醇的浓度的平方成正比,醇的浓度对硅消耗速率的贡献最大。
提高醇的浓度是提高反应速率即硅消耗速率的有效措施。
对于解离反应,只有解离了的醇能够参与反应。未解离的醇不能参与反应(4.1)。
因此,又有推论: 解离速度是反应速度的控制。提高醇的解离速度是提高反应速率的有效措施。
在甲醇-硅过程中,解离剂与催化剂同样重要。
9. 扩散过程与选择性控制
不管是硅的消耗速率,或者醇的消耗速率,
这二者的快速消耗,既可能是快速产生目标产物,也可能是快速产生副产物。
如何快速产生目标产物,而少产生或不产生副产物是我们追求的目标。
固体硅颗粒周围,存在一个由甲醇,
B2,B3,B4 和 B5 等物质构成的氛围。
甲醇向气体滞流膜内扩散,浓度逐渐形成一个沿半径方向向内降低的梯度。
反应产物解吸向外扩散形成一个由内向外沿半径方向降低的梯度。
向内传递的甲醇与解吸后向外传递的产物 B2 存在相遇的几率,会发生反应变成 B3 。
甲醇遇上 B3 会反应变成 B4 或 B5 。醇浓度越高,副反应越激烈。
因此,不能用加大醇浓度,追求硅的快速消耗。而应该寻求反应速率与效益的平衡优化。
在醇-硅过程中,主要矛盾是醇的转化不完全。关注对象是醇,而不是硅。
研究工作的视点应该放在醇和醇的转化率上,力求醇快速而完全地转化。
经验表明, B2,B3 与醇的反应不需催化,
也不需醇解离,控制醇与产物的返混率,是优化产物的选择性的关键。
决定醇转化为 B2 的条件是醇与硅进行化学反应的基本条件,如适宜的温度、压力、解离条件、催化剂活性和扩散传质速率等等。
反应(4.2)-(4.5)竞争消耗醇,既不需要催化,也不需要醇解离。而反应(4.1)既需要催化,又需要醇解离。
醇的解离速度是决定醇快速转化为 B2 的关键。如果反应(4.1)不能快速反应,则 B2 的产生在竞争中处于劣势,势必副产物增多。
相反,如果醇被快速转换掉了,副反应处于劣势,副产物会很少。所以,解离剂与催化剂的选择十分重要。
除此之外,决定醇完全转化的条件是固体物的吸附特性和气固两相充分的接触时间。
设反应器内固体粉末的表面总面积为 S,在特定条件下的吸附量是个常数,与具体的物质的吸附常数α有关,吸附量为 αS 。
经过一个吸附周期后,被吸附的气体完成一个化学吸附周期。
如果加料速度超过了αS,则多余的原料气会随流进入产物流中,增加副反应的几率。
表面面积、吸附常数和化学吸附周期是三个本质参数,确定了系统的效率和处理能力。吸附解离功在吸附剂,
增加比表面积的措施是减小粉末粒度,粒径越小,比表面积越大,越有利于提高吸附能力。
极性解离则功在溶剂,催化解离功在催化剂。高的溶剂极性和催化剂活性,都提高反应(4.1)的速率,降低其他反应消耗醇的竞争力。
这意味着这些措施有利于提高选择性,不利于副反应。
除了溶剂和催化剂,要提高选择性,关键在于控制返混率。返混率的控制靠的是反应器设计。
溶剂和催化剂是易耗品,价格昂贵,而设备是一次投资,突显了好的反应器设计的意义。
全混流中,物质浓度与位置无关,气体会迅速分散到整个空间,必然会有部分原料气进入到产物流中。
因此,全混流的选择性很低,不是复杂反应应当选择的反应器。
仅当目标产物在反应的较后步骤时,允许适当的返混率。幸运的是醇硅过程中,主产物是 B3,允许一定的返混率。
副反应步骤通常处在反应的较后步骤,这些反应步骤可以通过控制甲醇浓度来实现。
避免甲醇在固体料层中分布处于积累状态,可以降低副反应的概率。
原料气在反应器内的分布不可能那么均匀地给予每个硅颗粒正好一层。容易计算,如果只给一层吸附量,
物料馈送速率太低,生产效率太低。吸附与解吸动态地平衡,吸附量随机地分布,反应按概率在活性中心发生。
多种可以互相发生化学反应的气体混合物存在时,如果没有能完全抑制不需要的反应发生的措施,
其一种产物选择性不可能达到 100%。所谓选择性控制,指的是使反应有利于所需要的产物,而不利于副产物。
控制反应器内相关成分的比例,控制产物与原料气的混合比,可以提高选择性。
既实现原料气的完全转化又实现产物选择性控制,从原料气变成产物气必须按要求快速行动,即原料气在固体层中的扩散运动要快。
一旦化学反应完成之后变成了所需的产物就要迅速解吸并离开反应区(缩短化学吸附周期)和离开反应器,
降低与原料气不适当地再次相遇的概率。技术关键是设计出特定的化学工艺和反应装置,平推流或近似平推流反应器。
10. 气固反应器中的运动
醇-硅过程是一个复杂的化学吸附过程,不能只考虑反应(4.1)的行为。
扩散和吸附是过程,需要一定的时间,才能使原料气均匀扩散到整个空间中的每一颗粒的表面。
在这一段说起来很短的时间里会发生许多事情。
扩散从宏观向微观过渡。假定反应器是一个固定床(不带搅拌器),容积足够大,硅粉料层足够厚,
原料气经导气管进入反应器时,醇气体离开导气管时有一个初始流速。
在动量的推动下,气体在固体层中运动。这种运动受到固体的阻挡,速度逐渐降低,最后失去“流”的能力。
气体在固体层中的运动又服从扩散定律,从浓度高的地方向浓度低的地方扩散,最后透过气体滞留膜向颗粒表面扩散并被吸附。
假如给予的是一个长度足够短的气体脉冲。
可以想象,气体在料层中的扩散过程不是一开始就形成栓塞状的分配形式,而是树的形状。
“树”渐渐变得形影模糊并最后消失。逐渐弥漫扩散到整个反应器,达到平衡。
在扩散和吸附过程中,被吸附的气体在活性中心处发生化学反应,产物解吸向上运动。
一个气体脉冲在反应器内占据一段空间,成为一个栓塞,向上移动。在整个反应器内,
原料气的分布规律大体是从下部向上浓度逐渐降低,产物浓度则解吸向上流动形成相反的浓度梯度。
如果这棵树的树顶冲破了固体料层的顶界面,则部分原料气会直接进入产物流中。
当气源压力很高时,气流初速度很高,形成固体流态化现象,直接流向产物的原料气更多。
假如流动是理想的,甲醇和产物在硅粉固定床中的运动是等速的,
气体在固体层中的运动路径可以描述如图 10.1。
图 10.1 气体在固定床中的理想运动路径
假定在 t0时刻有 3 个可编号甲醇分子离开导气管进入床层中开始扩散。分别在三个不同时间被吸附住。
又分别停了时间t吸附和经过了一段时间 t反应 反应都变成了 B2 ,
再经过了一段时间t解吸后被解吸进入气体物流以同样的速度上升,同时在位置 C 相遇。
即同一时刻离开导气管的分子不管它们在什么位置上被吸附,最终聚集在同一高度位置上。
前面已经说过,扩散,吸附,反应,解吸,这些时间是些统计量,对每个分子不是完全相等的,那么相遇的位置可能有误差,这个误差是个微观量。
容易推理,一个时长 dt 的甲醇脉冲产生的产物经过一个吸附周期之后在反应器的顶端形成一个 dl 长的产物栓塞。
假如在 t1 时刻有第 4 号第 5 号等甲醇分子从导气管口流出,也将出现同一情形,
相对于第 1,2,3 号分子有一个时间差 t1-t0。
可以推断,若 t1-t0小于扩散周期,则前期被吸附的分子还没有完成化学吸附周期,还呆在原处。
第 4 号第 5 号等甲醇分子最终会遇上 t0 时刻进入硅粉层被吸附住的甲醇分子,
或其所产生的产物并发生继发反应步骤。后一时刻的气体大部分会沿着前一时刻的气体的路径更快地前进。
先是越过那些先已被吸附留下来的气体,然后冲到最前面形成新支流。
直到先前被吸附留下的气体完成化学吸附过程进入气体流中,
气流不再是单纯的原料气流,而是原料气与产物气的混合流了。这时便发生了原料气与产物的混合。
一部分原料气需要去补充已经反应解吸的空位,气体分子队列发生了紊乱。
因此,在固定床层中有吸附的流动不是平推流,而是蠕动。
相对而言,扩散比较慢而吸附和化学反应比较快。而宏观扩散过程在尺度上比较大,不是一个小量。
一个长度为 t1-t0 的原料气脉冲,其内部,产物与原料气会发生混合。
逐渐向上运动到反应器的顶部形成一个具有一定厚度的栓塞。
t1-t0 越小,栓塞厚度越簿。如果这个脉冲长度达到或超过吸附周期的长度。
则一个栓塞与另一个栓塞会有重叠,原料与产物的混合率会比较高。
11. 气固催化反应平推流实验模型
11.1 综述
下面讨论非全混流的系统设计。平推流装置的设计不容易,非全混流是所有平推流或近似平推流装置的总称。
这类设计可以有很多,以下讨论典型的四种,仅以此示意非全混流实验模型设计原理。
由这一原理,可以导出各种各样的平推流或近似平推流装置。
11.2 平推流反应器的概念设计
一种气固复杂反应的平推流反应器的概念如图 11.1 所示。
图11.1 平推流反应器示意图
反应器具有循环结构,固相物料及催化剂先放入加料斗(1)中,(2)为推进器旋转轴,
电机带动推进器旋转,将固相物料从加料斗(1)处吸入并沿箭头所指方向向前推进,
在原料气入口(3)处又吸入原料气,在混合扩散区完成混合扩散过程,被吸附,再被后来的物料推向前进,
进入化学反应过程,在产物减压解吸区完成解吸过程,
气体产物从产物排出口(5)排出反应器,未反应的固相物料沿(4)的方向沉降入固相物料加料斗,进入下一个周期。
只要原料气的流速恰当,该反应器内的流动为平推流。
如果反应物不是固体,而是液体,其产物为气体,可行性更好。
11.3 脉冲反应器
如图 11.2 所示,脉冲反应器主要由以下几部分组成:
- 反应器:一根直管,内部预装经过标准处理过的硅粉与催化剂的混合物。外包加热系统并测量反应器内温度的传感器。
下部为气体入口,气体经适当分配后进入料层,顶部设气固分离设备,产物气流向产品收集器;
- 原料储罐:气体或液体原料存储在储罐中,预热气化到指定压力待命;
- 脉冲阀:带脉冲发生器的电磁阀,一端连接原料气罐,常闭;出口连接反应器下部入口;
- 产物收集器: 产物经冷凝收集于收集器;
- 尾气收集袋: 尾气收集于尾气袋中。

图11.2 脉冲反应器示意图
脉冲流是最廉价的近似平推流,可以用来实验测定和估计某些工程参数并探讨平推流的效能。
常规控制。反应器内粉层高度,催化剂与硅粉的比例及活化方法,反应温度,反应压力,甲醇纯化处理方法,溶剂品种、处理方法及用量,等等。
测试项目。色谱测试:产物中甲醇% Me,三甲氧基硅烷%TMS,四甲氧基硅烷% TTMS。
估计:甲醇转换率MTP。必要时还可以增列其他考察项目。
输出 X :
- 脉冲周期长度 Pa,
- 单个脉冲长度Po(脉冲阀开启时间,Pa-Po为关闭时间),
- 醇气化压力 P(决定了脉冲强度),
- 是否惰性气体载流 G(氮气或氢气)及流量。
必要时还可以增加其他变量,例如,催化剂品种;溶剂品种等等。
11.4 螺旋式反应器
图11.3 螺旋式反应器示意图
这是一种平推流。反应器为一个两层结构,外层为一个标准的反应釜,中间为一个桶状结构,带一螺旋式搅拌器。
固体反应物予先加入釜内,加热到指定温度后,电机带动螺杆旋转,原料气从下面吸入,与固体物料混合,扩散,沿轴向提升向上行进。
完成反应后,在顶部解吸排出反应器,固体物料从反应器外层沉降循环。
反应器的内层与外层空间恰当配合以保证空间效益。反应器的内层长度 L 的高度以一个反应周期为度。
原料气的馈送速度与螺杆的旋转速度应该恰当配合,调节得使原料气进入到达顶端时,一个反应周期完成,或略有富余。
反应器设计完成后,实验参数实际上只有两个:原料气流速,和螺杆转速。
在同一配方体系下,系统的能力取决于内筒的容积、原料气流速和螺杆转速。
加快原料气流速和螺杆转速,则必须加长内筒的长度 L。如果解离剂及催化剂的效率更高,相应可以提高原料气流速和螺杆转速,从而提高系统的总效率。
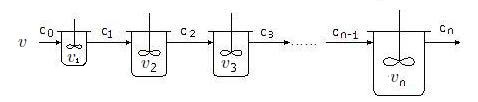
11.5 串联釜反应器
串联反应器是实现复杂反应的合适装置,如果需要加装其他物料如溶剂皆按容积比加入。
原料气进入一个全混釜,扩散会非常快,很快分散到该釜的每个角落。紧接着,原料气很快被均匀地吸附。
这是全混釜的优点。如果在该釜中实现了反应 (4.1),未被吸附的原料气汇合产物流进入反应器的顶端,排出该釜。
如果存在第二釜,第一釜排出的气体作为第二釜的输入,动作与第一釜类似,不同的是进入第二釜的气体是混合体,
体积流速比前一釜的入口小,减小的比例与产物的分布依赖于前一釜中的反应的速率和停留时间。
如果实现了完全的反应,体积减半。因此,第二釜的容积也应该减小,减小的比例依赖于第一釜的反应状态。
若采用连续流,前期被吸附的气体还没有完成其化学吸附周期,新的原料气接踵而至,
先期原料气完成其化学吸附周期之后,立即汇入原料流,成为混合流。
后期来到的原料气,一部分填补先期完成化学吸附周期解吸退出的空位,另一部分与先期的产物混合,返混没有避免。
幸而我们需要的产品是 B3,允许一定的返混。这就要求恰当的停留时间分布。
为了有效避免副产物,醇流应该是断续流或脉冲流。建议由恒流惰性气体载流脉冲醇气流。
这样,串联釜反应器是脉冲反应器的延伸。可以由脉冲反应器测定有关参数,然后设计串联反应器的级数,体积分布,产量和醇流速。
一个脉冲原料气进来,原料气很快被均匀地吸附。
多余的未被吸附的原料气流入第二釜,完成与第一釜同样的动作。完成其化学吸附周期之后,被推动前进。
载流惰性气体(如果有)将推动产物流,腾位给下一个脉冲原料气。这将是良好的流动。
原料气的转换率将非常高,副产物将非常少。
在脉冲反应器中测得的参数,在串联反应器上应该统调调整,以适应串联反应器。
如果串联反应器是全混的,载流是否还需要?需要实验决定。应该参考成本决定。
附录: 微分方程及其解
为书写方便和便于迭代计算,将硅简记作 B1 (不至误会时,有时也记作 B),
H2B(OR)2 为 B2, HB(OR)3 为 B3,
B(OR)4 为 B4, RB(OR)3 为 B5 。
在这一组记号下,假设反应器是一个容积足够大足以容纳全部原料和产物的全混釜。
将 M 摩尔硅粉及催化剂加入其中,并且假定每一个硅原子都以平等的身份参与反应。
以等速 v 摩尔/1分钟加入甲醇(ROH),且 v 适当大,使系统能维持 ROH 的浓度 CMe 为常值,直到反应完成。
5 个反应,相应有 5 个反应速率 k1,...,k5 ,需要实验测定。在时刻 t,各组分 Bi 的浓度变化,
从(4.1)-(4.5)可以得到微分方程组
dB1(t)/dt = -k1B1CMe2 ------(A.1)
dB2(t)/dt = -k1B1CMe2 -B2CMe ------(A.2)
dB3(t)/dt = -k2B2CMe-B3CMe(k4+k5) ------(A.3)
dB4(t)/dt = -k3B2CMe2 +k4B3CMe ------(A.4)
dB5(t)/dt = k5B3CMe ------ (A.5)
Bi 应当满足关系
B1+B2+B3+B4+B5=M ------(A.6)
t=0 时 Bi=0,(i=1,2,3,4,5) 。上述 5 个微分方程只有 4 个是独立的,满足关系
∑dBi/dt=0
在稳态操作条件下,假定甲醇的加料速度 v 适合使甲醇的浓度 CMe 是一常数,可并入 ki 中。
可以将(A.1)- (A.5)重写作
dB1(t)/dt = -k1B1 ------ (A.7)
dB2(t)/dt = -k1B1-B2 ------ (A.8)
dB3(t)/dt = -k2B2-B3(k4+k5) ------ (A.9)
dB4(t)/dt = -k3B2+k4B3 ------ (A.10)
dB5(t)/dt = k5B3 ------ (A.11)
这是一组线性非齐次常微分方程,逐个方程分离变量,应用常数变易法,积分结果如下。
B1(t) = c1exp(-k1t)
c1 = B1(0)=M
B2(t) = -c2exp(-k1t) + c2 exp(-k23t)
k23 = k2+k3
c2 = k1c1 / k23-k1
B3(t) = c31 exp(-k1t)+c32 exp(-k23t) +c3 exp(-k45t)
k45 = k4 + k5
c3 = -c31 -c32
c31 = k2k45-k1
c32=k2c2k45-k23
c41 = k4c313c2/-k1
c42=k3c2+k4c32/-k23
c43=k4c3+k4c32/-k45
c4 = - c41- c42- c43
vB4(t) = c41 exp(-k1t)+c42 exp(-k23t) + c43 exp(-k45t)+c4
c51 = k5c31 / -k1
c52= k5c32 / -k23
c53= k5c3 / -k45
c5 = - c51 - c52 - c53
B5(t) = c51 exp(-k1t) + c52 exp(-k23t) + c53 exp(-k45t) + c5
B5(t) 也可以由(A.6)式算出。
参 考
- 幸松民,王一璐,《有机硅合成工艺及其应用》,化学工业出版社,北京,2002
- (苏)涅克拉索夫著,张青莲等译,《无机化学教程》,北京,高等教育出版社,1953
- 严宣申,王长富,《普通无机化学》,北京大学出版社,1987
- 陈甘棠 主编,《化学反应工程》,化学工业出版社,北京,1981
- 黄恩才 主编,《化学反应工程》,化学工业出版社,北京,1998
- 南京大学物理化学教研组等编 ,《物理化学》,人民教育出版社,北京,1961
- 傅献彩,沈文霞, 姚天扬, 《物理化学》,(第四版),人民教育出版社,北京,1990
- 周公度,《结构化学基础》,北京大学出版社,(1989)
- (美)Charles N.Satterfield 著, 庞礼译, 《实用多相催化》,北京大学出版社,1990
- HANDBOOK OF CHEMISTRY AND PHYSICS 82ND (2001-2002)
- 烷氧基硅烷的直接合成工艺, ZL 02113594.0 (2002)
- 胡华明,胡文斌,李凤仪 直接合成三烷氧基含氢硅烷反应器分析, 《化工中间体》,2006年5月号, (2006)
- 杨春晖,张 磊,李 季,杨 恺,葛士彬,胡成发,直接法合成三烷氧基硅烷的研究进展,《有机硅材料》Vol.24(1),(2010)